文章目录
Install
(base) [yut@io02 01_Morchella]$ mamba create -n drep -c bioconda drep
依赖关系
dRep 需要其他程序才能运行。并非所有操作都需要所有依赖程序,要检查系统中安装了哪些依赖项,dRep 可以访问这些依赖项,请运行
(drep) [yut@io02 01_Morchella]$ dRep check_dependencies
mash.................................... all good (location = /datanode02/yut/Software/Miniconda3/envs/drep/bin/mash)
nucmer.................................. all good (location = /datanode02/yut/Software/Miniconda3/envs/drep/bin/nucmer)
checkm.................................. !!! ERROR !!! (location = None)
ANIcalculator........................... !!! ERROR !!! (location = None)
prodigal................................ all good (location = /datanode02/yut/Software/Miniconda3/envs/drep/bin/prodigal)
centrifuge.............................. !!! ERROR !!! (location = None)
nsimscan................................ !!! ERROR !!! (location = None)
fastANI................................. !!! ERROR !!! (location = /datanode02/yut/Software/Miniconda3/envs/drep/bin/fastANI)
- 安装其他依赖
# install
$ mamba install -c bioconda checkm-genome
# download database
$ wget https://data.ace.uq.edu.au/public/CheckM_databases/checkm_data_2015_01_16.tar.gz
$ [/datanode02/yut/Database/checkm_data_2015_01_16] tar -zxvf checkm_data_2015_01_16.tar.gz
(drep) [yut@node02 checkm_data_2015_01_16]$ checkm data setRoot /datanode02/yut/Database/checkm_data_2015_01_16
[2023-11-10 20:06:35] INFO: CheckM v1.2.2
[2023-11-10 20:06:35] INFO: checkm data setRoot /datanode02/yut/Database/checkm_data_2015_01_16
[2023-11-10 20:06:35] INFO: CheckM data: /datanode02/yut/.checkm
[2023-11-10 20:06:35] INFO: [CheckM - data] Check for database updates. [setRoot]
Path [/datanode02/yut/Database/checkm_data_2015_01_16] exists and you have permission to write to this folder.
(re) creating manifest file (please be patient).
Path [/datanode02/yut/Database/checkm_data_2015_01_16] exists and you have permission to write to this folder.
(re) creating manifest file (please be patient).
常用命令
输入文件可以是.fna或者fna.gz或者混合的
(gtdbtk) [yutao@globin test]$ ls *fna*
fna.fa GCA_000685155.1_ANME2D_V10_genomic.fna.gz test.fna
(gtdbtk) [yutao@globin test]$ time dRep dereplicate drep_out --ignoreGenomeQuality -p 20 -g *fna*
(gtdbtk) [yutao@globin test]$ cat drep_out/data_tables/genomeInformation.csv
location,length,N50,genome,centrality
/home/yutao/test/fna.fa,3203386,545726,fna.fa,0.0
/home/yutao/test/GCA_000685155.1_ANME2D_V10_genomic.fna.gz,3203386,545726,GCA_000685155.1_ANME2D_V10_genomic.fna.gz,0.0
/home/yutao/test/test.fna,199347,46542,test.fna,0.0
(drep) [u@h@3241TGG_High_Quality_Genomes]$ nohup time dRep dereplicate 3241Genomes_dRep_0.99 -comp 0 -con 100 -p 28 -sa 0.99 -g 3241TGG_genomes/*.fa &>drep_0.99.log&
(drep) [u@h@GEM_env_sets]$ nohup dRep dereplicate GEM_ENV_dRep0.95 --ignoreGenomeQuality -p 28 -sa 0.95 -g *.fa &>gem0.95.log&
常见问题
pplacer线程超过30报错
dRep v3.2.2 超过30线程,pplacer进程一直在,但是没有计算结果。
当比较基因组很多(>4096)
当比较基因组很多时,某些系统会限制参数打开个数,需要将基因组的路径写到一个文件,跟在-g
(gtdbtk) [yutao@myosin release202]$ find $PWD/tmp -iname "*fna*" -type f >48862genome_path.tsv
(gtdbtk) [yutao@myosin release202]$ nohup time dRep dereplicate 968OTU_vs_RS202_47894OTUs_dRep0.95 --ignoreGenomeQuality -p 80 -sa 0.95 -g 48862genome_path.tsv &>drep.log&
- 注意:一定要将路径写对,有一个不对,则会导致checkM错误
有了Bdv.csv文件后无需输入基因组list
如果有了Bdv.csv文件,则w无需输入基因组list,直接去掉-g选项及参数
超多基因组
使用--ignoreGenomeQuality -p 150 --S_algorithm fastANI --multiround_primary_clustering --greedy_secondary_clustering快速比较5000+基因组
(gtdbtk) [yutao@globin GCS_OTUs_vs_OtherDB]$ nohup time dRep dereplicate GCSvsGEM_FastANI_dRep95 --ignoreGenomeQuality -p 100 --S_algorithm fastANI --multiround_primary_clustering --greedy_secondary_clustering -sa 0.95 -g 47516_GEM.path &>47516_GEM_fastANI.drep.log &
# 近48k基因组耗时69h
# 可通过以下命令查看进度
$ grep -c "running cluster" GCSvsGEM_FastANI_dRep95/log/logger.log
39141 #已经聚类的数量
$ grep "primary clusters made" GCSvsGEM_FastANI_dRep95/log/logger.log
10-25 05:54 INFO 39141 primary clusters made # 总的二级聚类数量
nohup time dRep dereplicate GCSvsGEM_dRep95 --ignoreGenomeQuality --multiround_primary_clustering -p 80 -sa 0.95 -g 47516_GEM.path &>47516_GEM_drep95.log &
# 不做checkM,超过30核
# 耗时超过6天未出结果(256threads & 500G),因为secondary cluster是成对的并且不是使用FastaANI,非常慢
- 线程大于30的时候,
--ignoreGenomeQuality要放到最前面,否则报错
为什么需要界定种?
在许多情况下,确定微生物之间的关系是研究问题的中心。 居住在建筑物表面的微生物是否与居住在其租户中的微生物相同? 医院病房中的微生物是否与新生婴儿中的微生物相同? 生活在木制物表面的大肠杆菌与生活在塑料的大肠杆菌一样吗?常常通过平均核酸相似性(Average Nucleotide Identity, ANI)来衡量。 基本思想是比对两个基因组并计算比对中错配的数量。 例如,ANI为99%的基因组每100个碱基之间有1个错配,而ANI为95%的基因组每100个碱基之间有5个错配,依此类推。 有许多计算平均核苷酸同一性的方法,主要区别在于用于比对基因组的算法差异。2–4
通过计算许多系统中基因组之间的ANI,已记录了一些松散的和一般的ANI断点:
- < 96% ANI = Same 16S cluster (using standard 97% clustering)5
- 96% ANI = Same bacterial species4
- 98% ANI = Same E. coli clade6
- 98.8% ANI = Same Prochlorococcus clade7
- 99.9% ANI = Same K. pneumoniae outbreak strain8
在哪个ANI阈值处将基因组称为“相同”更为合理,这取决于研究问题。 如果问题是弗拉格斯塔夫办公室中的微生物与圣地亚哥办公室中的微生物是否相同,那么属于同一个species微生物即可,因此ANI为95%(或16S测序) )将足以解决这个问题(确实如此; Chase,20169)。 如果问题是两个不同身体部位的微生物是否来自同一来源,那么95%ANI太低了。 仅仅因为大肠杆菌位于两个身体部位并不意味着它们来自同一地点; 一种菌株可能来自土壤,另一种菌株来自隔壁的邻居。 绝对需要95%以上的ANI才能显示两种菌株都来自同一来源,但是到底需要多高的ANI这其实是另一个问题(在最近的出版物中使用99.9%的ANI来解决这个问题; Olm,201610)。
为特定问题选择ANI阈值时,可视化基因组之间的关系通常会很有帮助。 dRep是最近在bioRxiv11上发布的python程序,旨在实现这一目的。 例如:

上图显示了匹兹堡同一重症监护室中居住在不同婴儿中的链霉菌(Streptomyces )菌株之间的ANI。 从图中可以看到,conN3_174_037G1_concoct_13和conN1_023_029G1_concoct_18之间的ANI约为99.25,conN3_174_023G1_concoct_19和conN3_174_021G1_concoct_4之间的ANI约为100,依此类推。 该图还清楚地表明,不同的ANI阈值将得出关于哪些婴儿具有“相同”菌株的不同结论。 例如,如果其ANI> = 98.5%(如黑色虚线所示),则称基因组为“相同”(same),将得出结论,即所有婴儿都只有一个链霉菌菌株。 如果其ANI> = 99.5%(如红色虚线所示),则称基因组为“相同”,将得出结论,有4个不同的链霉菌菌株(上面2个可视为同一种,中间三个可视为同一种;最下面2个是2个种),其中两个(conN3_174_037G1_concoct_13和conN1_023_029G1_concoct_18)同在一个婴儿中。 在此示例中,将ANI阈值更改单个百分点会完全更改从数据得出的结论,从而突出显示了谨慎选择阈值的重要性。
- dRep的大致流程

- 1.Assemble each sample separately using your favorite assembler. You can also perform a co-assembly to catch low-abundance microbes
- 2.Bin each assembly (and co-assembly) separately using your favorite binner
- 3.Pull the bins from all assemblies together and run dRep on them
- 4.Perform downstream analysis on the de-replicated genome list
dRep_114">dRep重要概念
在使用dRep进行基因组去重复和基因组比较的时候,需要理解以下几个重要概念:
- 1.选择合适的次级ANI阈值:这个阈值规定了基因组需要有多相似才能被认为是相同的。本节将介绍如何选择适当的
S_ANI阈值; - 2.最小比对覆盖率。在计算基因组之间的ANI时,我们还确定了基因组中进行比较的覆盖度(fraction)。该值可用于建立基因组之间所需的最小重叠水平(
cov_thresh),但在使用此参数时需要注意很多问题; - 3.选择具有代表性的基因组。在去重复过程中,第一步是识别相似基因组的类别,第二步是为每个类别选择一个代表性基因组(RG);
- 4.使用贪婪算法。贪心算法是走捷径来获得解的算法。dRep可以在几个阶段使用贪婪算法来加快运行速度;本节将介绍如何使用它们以及需要注意什么;
- 5.基因组完整性的重要性。由于它对Mash基因组的依赖,dRep必须在一定程度上完成对它们的处理。本节解释原因;
- 6.奇怪的分层聚类。 在比较所有基因组之后,dRep使用分层聚类将成对的ANI值转换为基因组簇。本节介绍了这件事以及它可能出错的地方;
- 7.基因组比较算法概述。dRep有很多不同的基因组比较算法可供选择。本节简要介绍它们的工作原理和它们之间的区别;
- 8.比较和去重复非细菌基因组。在比较噬菌体、质粒和真核生物时,一些关于使用适当参数的想法;
次级ANI的选择
1.对于两个 "相同 "的基因组之间共享的平均核苷酸身份(ANI),没有一个定义。这是一个用户必须根据自己的具体应用情况自行决定的。ANI是由二级聚类算法决定的,最小二级ANI是被认为是 "相同 "的基因组之间的最小ANI,最小对齐部分是相信报告的ANI值的最小基因组重叠量。见7. 基因组比较算法概述中关于二级聚类算法的描述和2. 关于调整最小对齐分数的概念,见7.最小对齐覆盖率。基因组去重复的一个用例是把一大群基因组拿出来,挑出一套物种级的代表基因组(SRG)。最近,Almeida等人使用dRep完成了这项工作,其目的是为生活在人类肠道中的微生物产生一套全面的高质量参考基因组。对于划分不同物种的细菌的确切阈值存在争议,但大多数研究都认为95%的ANI是物种水平去重的适当阈值。关于如何选择这个阈值的背景,以及关于细菌物种在自然界中是作为一个连续体还是作为离散单位存在的一些争论,见Olm等人,2020。去重复的另一个用途是产生一组足够不同的基因组,以避免错误的映射。如果我们将元基因组读数(通常是~150bp)映射到两个99.9%相同的基因组上,大多数读数将同样映射到两个基因组上,而你将无法分辨哪个读数真正 "属于 "一个基因组或另一个。如果去重复的目标是在映射短读数时产生一组不同的基因组,那么98%的ANI是一个合适的阈值。关于98%的数值是如何得出的,请看下面的页面。
dRep中的默认值是99%,这是对二级ANI阈值的精确程度的限制。作为一个经验法则,阈值高达99.9%可能是安全的,但高于这个值就超出了检测的极限。将基因组与自身进行比较,通常会产生~99.99%的ANI值,因为算法并不完美,会被基因组重复区域和类似的东西甩开。参见InStrain程序的出版物,了解有关测试dRep检测极限的细节,以及如果需要的话,如何进行更详细的染色剂水平比较。
Minimum alignment coverage
最小对齐分数是基因组必须重叠的最小数量,以使报告的ANI值 “计数”。这个值是作为ANIm/gANI算法的一部分报告的。
想象一下这样的情景:两个远缘的基因组共享一个相同的转座子。当基因组比较算法运行时,转座子可能是基因组中唯一对齐的部分,并且对齐后的ANI值为100%。这将导致报告的ANI为100%,而报告的对齐率为~0.1%。最小对齐分数是为了处理上述情况–任何少于最小对齐分数的基因组对齐都会使ANI变为0。
dRep处理最小对齐分数的方式可以说明上述情况,但总体上是非常笨拙的。当一个比较低于最小对齐分数时,dRep实际上只是将ANI从算法报告的任何内容改为 “0”。这可能会给接下来的分层聚类带来问题(见7.基因组比较算法的概述)。我试着思考更好的处理方法,但这是一个难以解释的问题。你如何处理一组在20%的基因组上共享100% ANI的基因组?然而,在使用dRep多年后,我得出这样的结论:在实际情况下,这很少是一个问题。在大多数情况下,对齐的部分要么非常高(>50%),要么非常低(>10%),所以10%的默认值在大多数情况下是有效的。参见Olm等人2020年的图1,描述了典型的物种内(基因组共享>95% ANI)的比对覆盖率值,注意>95% ANI的基因组几乎不会有低比对覆盖率。
有人建议,物种级别的分类定义应该采用最低60%的对齐比例(Varghese 2015)。然而,当使用不完整的基因组时,这可能太严格了(正如基因组去重复时经常出现的情况)。
3. 选择有代表性的基因组
dRep使用一个基于分数的系统来挑选代表基因组。聚类中的每个基因组都被赋予一个分数,分数最高的基因组被选为代表。这个分数是基于以下公式。
A∗Completeness−B∗Contamination+C∗(Contamination∗(strainheterogeneity/100))+D∗log(N50)+E∗log(size)+F∗(centrality−Sani)
其中A-F是命令行参数,默认值分别为1、5、1、0.5、0和1。调整A-F可以让你决定在选择代表性基因组时对特定特征的权重。例如,如果你真的关心有低污染和高N50,你可以增加B和D。
完整性、污染性和菌株异质性由用户提供或用checkM计算。N50是衡量构成基因组的片段有多大。大小是基因组的总长度。中心度是衡量一个基因组与它的群组中所有其他基因组的相似程度。这个指标有助于挑选与所有其他基因组相似的基因组,并避免挑选相对离群的基因组。
一些出版物在挑选有代表性的基因组时,在其评分中加入了其他指标,如基因组是否来自分离物。例子见《人类肠道微生物组204,938个参考基因组的统一目录》和《细菌和古细菌的完整领域-物种分类法》。
4. 使用贪婪的算法
通俗地说,贪婪算法是那些走捷径的算法,运行速度更快,得出的解决方案可能不是最优的,但却 “足够接近”。贪婪算法越好,最优解和贪婪解之间的差异就越小。由于成对比较迅速扩展到需要数十年计算的水平,dRep使用一些贪婪算法来加速。
dRep使用的一种贪婪算法是初级聚类。执行这一步骤可以极大地减少必须进行的基因组比较的数量,减少运行时间。这样做的代价是,如果基因组最终出现在不同的主聚类中,它们将永远不会被比较,因此也永远不会出现在相同的最终聚类中。这就是为什么下面的章节(基因组完整性的重要性)很重要。
在2021年(dRep v3),引入了几个额外的贪婪算法,描述如下。这些都是相对较新的功能,所以如果你发现问题或有建议,请不要犹豫地联系我们。
-multiround_primary_clustering在一系列的组中进行初级聚类,然后在最后用单链路聚类进行合并。这极大地减少了RAM的使用,提高了速度,但代价是精度略有损失,而且不能绘制primary_clustering_dendrograms。在对5000多个基因组进行聚类时尤其有帮助。
-greedy_secondary_clustering在进行二次聚类时使用启发式方法来避免成对比较。其工作方式是,随机选择一个基因组来代表一个聚类。然后将下一个基因组与该基因组进行比较。如果它与该基因组的ANI阈值低,它将被放入该群组。如果不是,它将被放入一个新的集群,并成为新集群的代表基因组。然后,第3个基因组将与所有集群的代表进行比较,以此类推。这基本上导致了单链路聚类,而不需要进行成对的比较。不幸的是,由于FastANI需要不断地重新绘制基因组图谱,这并没有像你期望的那样提高速度。这个选项目前只适用于FastANI的S_algorithm。
-run_tertiary_clustering不是一种贪婪的算法,而是一种处理贪婪算法所带来的潜在不一致的方法。一旦聚类完成并选择了有代表性的基因组,这个选项就会对最终的基因组集再运行一轮聚类。这在进行贪婪聚类和/或处理类似的基因组最终出现在不同的主聚类中的情况时特别有用。这基本上是一个检查,以确保所有的基因组都是基于给定的参数而彼此不同的。
5. 基因组完整性的重要性
这个决定要比前者复杂得多。从本质上讲,在计算效率和最小基因组完整性之间存在着一种权衡。

图A:五个基因组被细分为10%-100%的部分,并对同一基因组的部分进行比较。X轴是允许的最小基因组完整度。这个值越宽松,对齐的分数范围就越大。
如上图A所示,基因组完整性的极限越低,两个基因组可能对齐的部分就越低。这是有道理的–如果你随机抽取一个基因组的20%,然后再做同样的事情,当你比较这两个随机的20%的子集时,你不会期望它们中有多少是对齐的。当你考虑到它对Mash的影响时,这个 "对齐的部分 "就真正成为一个问题。

图B: 一个相同的大肠杆菌基因组被子集到10%-100%的分量,并对分量进行比较。当较低数量的基因组对齐时(由于不完整),Mash ANI会受到严重的影响
如上图B所示,对于相同的基因组,对齐的部分越低,报告的Mash ANI越低。
请记住–基因组首先用Mash分为一级簇,然后每个一级簇被分为 "相同 "基因组的二级簇。因此,符合 "相同 "定义的基因组必须最终出现在同一个主簇中,否则程序永远不会意识到它们是相同的。由于更多的不完整的基因组具有较低的Mash值(即使这些基因组是真正相同的;见图B),你允许进入你的基因组列表的不完整的基因组越多,你必须减少主簇的阈值。
有一个较低的主集群阈值,这将导致更大的主集群,这将导致更多需要的二级比较。这将导致更长的运行时间。
还听我的吗?
例如,假设我将最低基因组完整性设置为50%。如果我取一个大肠杆菌基因组,将其50%的基因组子集2次,然后将这2个子集基因组放在一起比较,Mash将报告96%的ANI。因此,主聚类阈值必须至少为96%,否则这两个基因组可能最终处于不同的主聚类中,因此永远不会有二级算法在它们之间运行,因此也就不会被去重。
然而,你不想把主集群的阈值设置得过低,因为这将导致更多的基因组被包括在每个主集群中,从而导致更多的二级比较(这很慢),因此运行时间也会更高。
把这些放在一起,我们可以得到一个数字,即相同的基因组被子集到不同部分的最低报告ANI。这个数字只考虑到了5个不同的基因组,但给出了一个大致的限制概念。

最后要考虑的是,当真正运行dRep时,用户实际上并不知道他们的基因组有多不完整。他们必须依靠单拷贝基因清单这样的指标来告诉他们。这就是dRep目前不支持噬菌体和质粒的原因–没有办法知道它们有多完整,因此也没有办法过滤掉那些太不完整的分类。不过总的来说,checkM在访问基因组的完整性方面是非常好的。

挑选基因组完整度阈值的一些一般准则。
不建议低于50%的完整度。所得到的基因组无论如何都是非常糟糕的,甚至二级算法在这一点上也会崩溃。
降低二级ANI应该导致MASH ANI的全面降低。这是因为你想让Mash将非相似和不完整的基因组分组。
7. 基因组比较算法概述
一级聚类总是用Mash进行;这是一种极快但有些不准确的算法。
有几种支持的二级聚类算法。这些算法计算出基因组之间准确的平均核苷酸身份(ANI),用于将基因组聚成二级聚类。目前,从第3版开始支持以下算法。
ANIn(Richter 2009)。这是用nucmer对整个基因组进行比对,并对比对的区域进行比较。
ANImf (DEFAULT)。这与ANIn相同,但对排列进行过滤,使基因组1的每个区域只与基因组2的一个区域对齐。这需要稍多的时间,但在有重复区域的基因组上更准确。
gANI(Varghese 2015)。这是对Prodigal调用的基因(ORFs)进行对齐,而不是对整个基因组进行对齐。这个算法比基于ANIm的算法快一点,但只对齐编码区。
goANI。这是我自己对gANI的开源实现,gANI是不开源的(被问及时作者也不会分享源代码)。我写了这个算法,以便我可以为这项研究计算对齐的基因之间的dN/dS(你也可以使用dnds_from_drep.py)。需要程序NSimScan。
FastANI(Jain 2018)。一个真正快速的基于Mash的算法,也可以处理不完整的基因组。似乎与基于对齐的算法一样准确。当你关心运行时间的时候,可能应该是默认的算法。
这些算法都不是完美的,特别是在容易重复的基因组中。基因组中非同源的区域可以相互对齐,人为地降低ANI。事实上,当一个基因组与它自己进行比较时,由于这个原因,这些算法经常报告的数值<100%。
8. 对非细菌基因组进行比较和去重复
dRep是针对细菌解重复的使用情况而开发的,在非细菌实体上运行时,有一些事情需要注意。
需要注意的一个主要问题是初级聚类。正如在5.基因组完整性的重要性中所描述的,基因组需要>50%的完整性才能使初级聚类起作用。如果你正在比较那些你无法评估完整性的实体,或者你想比较那些只共享有限数量基因的基因组(如噬菌体或质粒),这就是一个问题。最简单的处理方法是用参数-SkipMash完全避免一级聚类,或者用-pa降低一级聚类的阈值。
还要考虑对准覆盖率(2.最小对准覆盖率)对分层聚类的影响(6.分层聚类的怪异性)。如果你的工作对象是那些特别具有镶嵌性的实体,如噬菌体,这可能是一个比细菌更大的问题。
基因组的过滤和评分也是一个主要因素。如果你的基因组不能被checkM评估,你可以用标志-ignoreGenomeQuality关闭质量过滤以及在挑选基因组时使用完整性和污染性。
在为我自己的研究(Olm 2019和Olm 2020)考虑这些选项时,我采用了以下dRep命令来对噬菌体和质粒进行聚类。请把这看作是一个人为他们的具体使用情况处理这些参数的尝试,不要害怕在你认为合适的时候做额外的调整。
dRep dereplicate --S_algorithm ANImf -nc .5 -l 10000 -N50W 0 -sizeW 1 --ignoreGenomeQuality --clusterAlg single
dRep_204">dRep结果处理
1.获得OTU members
awk -F, 'NR==FNR{a[$1]=$1}NR>FNR{if($1 in a ){print a[$1]"*,"$0}else{print $1","$0} }' Wdb.csv Cdb.csv >OTUs_members_Relationship.csv
第一列为所有基因组名称,代表基因组用星号表示,第三列为OUT id,相同id表示同一个OTU

2.比较两种生境基因组ANI距离分布密度图
Mdb.csv的第4列为两个基因组之间的相似性,随后基于基因组metadata信息,仅保留两个生境之间基因组的比较结果,用于绘制密度图
# Mdb.csv示例
(base) [yutao@myosin data_tables]$ head Mdb.csv
genome1,genome2,dist,similarity
RS_4.bin.50.fa,RS_4.bin.50.fa,0.0,1.0
8_GM_sbin_oily_55.fa,RS_4.bin.50.fa,1.0,0.0
SRR13892588_me2_bin.130.fa,RS_4.bin.50.fa,1.0,0.0
7_SB_sbin_7_41.fa,RS_4.bin.50.fa,1.0,0.0
HTR7_me2_bin.71.fa,RS_4.bin.50.fa,1.0,0.0
RS_1.bin.2.fa,RS_4.bin.50.fa,1.0,0.0
# 包含两种生境的基因组metadata示例
$ sed -i -e "s/_genomic.fa//g;s/.fa//g" Mdb_processed.csv
# 存在AB及BA的情况
(base) [yutao@myosin data_tables]$ awk -F"," '$1=="RS_4.bin.50" && $2=="2_HM_cbin_9"' Mdb_processed.csv
RS_4.bin.50,2_HM_cbin_9,1.0,0.0
(base) [yutao@myosin data_tables]$ awk -F"," '$2=="RS_4.bin.50" && $1=="2_HM_cbin_9"' Mdb_processed.csv
2_HM_cbin_9,RS_4.bin.50,1.0,0.0
# 保留其中一个即可
#echo -e 'a,b\nb,a\na,c'|awk -F"," '{if(!a[$1","$2]&&!a[$2","$1])a[$1","$2]=1}END{for(i in a){if(a[i])print i}}'
$ awk -F"," 'NR==1{print}{if(!a[$1","$2","$3","$4]&&!a[$2","$1","$3","$4])a[$1","$2","$3","$4]=1}END{for(i in a){if(a[i])print i}}' Mdb_processed.csv >Mdb_dereplicate.csv
(gtdbtk) [yutao@myosin data_tables]$ wc -l Mdb_processed.csv Mdb_dereplicate.csv
9180901 Mdb_processed.csv
4591967 Mdb_dereplicate.csv
# 添加分组信息,选择不在同一个组,且相似性值不为0的结果
(base) [yutao@myosin data_tables]$ awk -F"\t" 'BEGIN{print "MAG1,group1,MAG2,group2,dist,similarity"}NR==FNR{a[$1]=$2}NR>FNR{FS=",";OFS=",";if($1!~/genome/)print $1,a[$1],$2,a[$2],$3,$4}' MAGs_metadata.tsv Mdb_dereplicate.csv|awk -F, '$2!=$4 && $NF!=0'|head
MAG1,group1,MAG2,group2,dist,similarity
MAG_1907NJYTS1_metabat2_bin.13,Glacier,RS_4.bin.50,Cold seep,0.295981,0.704019
SRR5275918_metabat2_bin.57,Glacier,RS_4.bin.50,Cold seep,0.295981,0.704019
ISO_B518-1,Glacier,RS_4.bin.50,Cold seep,0.295981,0.704019
ISO_GM2-5-1,Glacier,RS_4.bin.50,Cold seep,0.295981,0.704019
MAG_KQGRI2.20.08_metabat2_bin.34,Glacier,8_GM_sbin_oily_55,Cold seep,0.295981,0.704019
MAG_18PLSC_vamb_S1C8874,Glacier,8_GM_sbin_oily_55,Cold seep,0.295981,0.704019
GCA_015751965.1_ASM1575196v1,Glacier,SRR13892588_me2_bin.130,Cold seep,0.295981,0.704019
ISO_B1382-2,Glacier,SRR13892588_me2_bin.130,Cold seep,0.295981,0.704019
ISO_N1997-1-1,Glacier,SRR13892588_me2_bin.130,Cold seep,0.295981,0.704019
# 或可在最后去重
(gtdbtk) [yutao@myosin data_tables]$ awk -F"," '{if(!a[$1","$3]&&!a[$3","$1])a[$1","$3]=1}END{for(i in a){if(a[i])print $0}}' GGG_and_CSMD_ANI_similarity.csv
根据drep ANI 0.99的聚类结果文件dRep_0.99/data_tables/Widb.csv的第11列cluster_members绘制Pie图
- Cdb.csv包含了每个OTU下的isolates
- 原始数据:dRep_0.99/data_tables/Widb.csv
[u@h@data_tables]$ head Widb.csv
genome,score,completeness,contamination,strain_heterogeneity,size,N50,cluster,taxonomy,tax_confidence,cluster_members,closest_cluster_member,furthest_cluster_member,completeness_metric,contamination_metric
MAG_1611DDSC2_vamb_S1C744.fa,1.7918826841424995,NA,NA,NA,NA,NA,1_1,NA,NA,2,100.00,100.00,NA,NA
MAG_1908TGLC3_vamb_S1C1195.fa,1.9215229052672849,NA,NA,NA,NA,NA,1_2,NA,NA,11,100.00,99.85,NA,NA
MAG_1807DKMD2_vamb_S1C96.fa,1.9449308606290945,NA,NA,NA,NA,NA,2_1,NA,NA,5,99.44,99.39,NA,NA
- 查看同一个OTU下的基因组来源
[yut.yut-PC] ➤ awk '{print $2}' tab.txt |sort -u|while read id; do type_num=$(awk -v id=$id '$2==id{split($1,a,"_");print a[1]}' tab.txt|sort -u|wc -l); otu_num=$(awk -v id=$id '$2==id' tab.txt|wc -l);if [ $type_num -eq 2 ];then printf $id"\t"$type_num"\t"$otu_num"\n";fi ;done
OTU212 2 21
OTU229 2 4
OTU259 2 7
OTU292 2 2
OTU349 2 16
OTU358 2 3
OTU359 2 7
OTU395 2 3
work directory file-tree
workDirectory
./data
...../checkM/
...../Clustering_files/
...../gANI_files/
...../MASH_files/
...../ANIn_files/
...../prodigal/
./data_tables
...../Bdb.csv # Sequence locations and filenames
...../Cdb.csv # Genomes and cluster designations
...../Chdb.csv # CheckM results for Bdb
...../Mdb.csv # Raw results of MASH comparisons
...../Ndb.csv # Raw results of ANIn comparisons
...../Sdb.csv # Scoring information
...../Wdb.csv # Winning genomes
...../Widb.csv # Winning genomes' checkM information
./dereplicated_genomes
./figures
./log
...../cluster_arguments.json
...../logger.log
...../warnings.txt
- 其他输出结果
gtdbtk) [yutao@globin F_Public_Database]$ ls 6023_MIMAG_MAGs_dRep95
data # 中间结果
data_tables # 数据统计结果
dereplicated_genomes # 去冗余后的基因组
figures # pdf图
log
# (gtdbtk) [yutao@globin 6023_MIMAG_MAGs_dRep95]$ ls data
ANImf_files # NUCMER 运行的每个基因组和相近基因组的delta.filtered结果
Clustering_files # 聚类的pickle文件secondary_linkage_cluster_1000.pickle
MASH_files # mash 结果
prodigal # prodigal预测结果,包含fna,faa,但不是所有基因组的
关于代表基因组选择
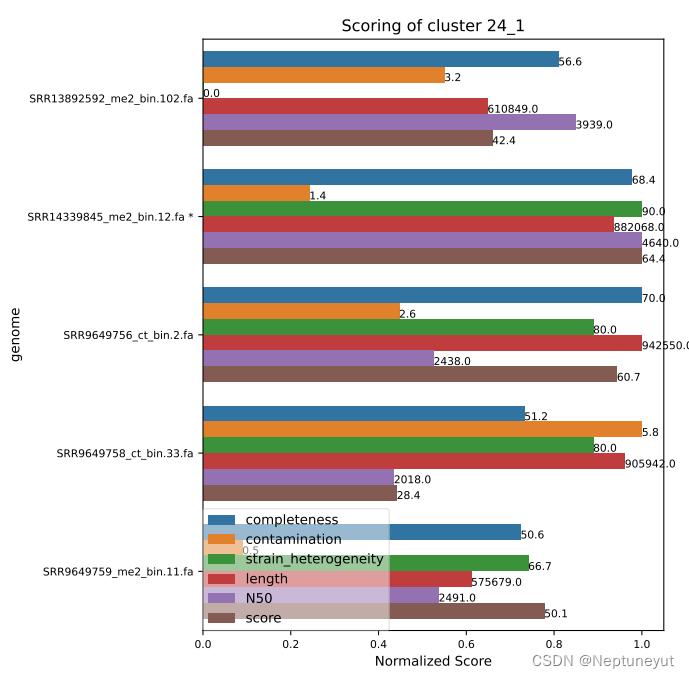
dRep在使用checkM确定基因组质量以后,会选择基因组完整度/污染度最优的基因组(Qality score=comp-5cont)作为代表序列,在cluster_score.pdf可以看到每个聚类及质量情况。
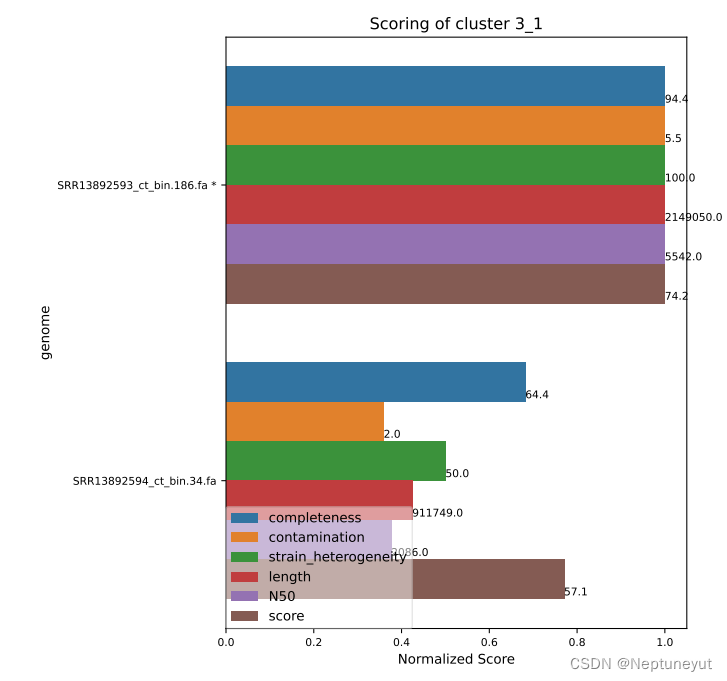
*代表OTU
使用
基因组去重复是识别基因组集合中“相同”的基因组,并从每个冗余集合中除去“最佳”基因组之外的所有基因组的过程。
测试去重复
两个基因组相似性0.98

--S_algorithm fastANI
-p PROCESSORS, --processors PROCESSORS
threads (default: 6)
--ignoreGenomeQuality
Don't run checkM or do any quality filtering. NOT
RECOMMENDED! This is useful for use with
bacteriophages or eukaryotes or things where checkM
scoring does not work. Will only choose genomes based
on length and N50 (default: False)
--S_algorithm {goANI,fastANI,ANImf,ANIn,gANI}
Algorithm for secondary clustering comaprisons:
fastANI = Kmer-based approach; very fast
ANImf = (DEFAULT) Align whole genomes with nucmer; filter alignment; compare aligned regions
ANIn = Align whole genomes with nucmer; compare aligned regions
gANI = Identify and align ORFs; compare aligned ORFS
goANI = Open source version of gANI; requires nsmimscan
(default: ANImf)
-pa P_ANI, --P_ani P_ANI
ANI threshold to form primary (MASH) clusters
(default: 0.9)
-sa S_ANI, --S_ani S_ANI
ANI threshold to form secondary clusters (default:
0.99)
(drep) [u@h@test]$ tree -L 2 drep_out/
drep_out/
├── data
│ ├── ANImf_files
│ ├── Clustering_files
│ └── MASH_files
├── data_tables
│ ├── Bdb.csv
│ ├── Cdb.csv
│ ├── genomeInformation.csv 所有基因组信息,N50,Size以及CheckM评估结果(若执行checkm)
│ ├── Mdb.csv
│ ├── Ndb.csv
│ ├── Sdb.csv
│ ├── Wdb.csv
│ └── Widb.csv
├── dereplicated_genomes 去重后的基因组
│ └── Ecoli_k12.fasta
├── figures 相关图片
│ ├── Clustering_scatterplots.pdf
│ ├── Cluster_scoring.pdf
│ ├── Primary_clustering_dendrogram.pdf
│ ├── Secondary_clustering_dendrograms.pdf
│ ├── Secondary_clustering_MDS.pdf
│ └── Winning_genomes.pdf
└── log
├── cluster_arguments.json
├── logger.log
└── warnings.txt
- de-replication
dRep dereplicate -p 20 outout_directory -g path/to/genomes/*.fasta
# 设置完整度/污染度
dRep dereplicate -p 50 -comp 0 -con 100000 dRep_out -g metabat2_bins/*fa
positional arguments:
work_directory Directory where data and output
*** USE THE SAME WORK DIRECTORY FOR ALL DREP OPERATIONS ***
-p PROCESSORS, --processors PROCESSORS
threads (default: 6)
-l LENGTH, --length LENGTH
Minimum genome length (default: 50000)
-comp COMPLETENESS, --completeness COMPLETENESS
Minumum genome completeness (default: 75)
-con CONTAMINATION, --contamination CONTAMINATION
Maximum genome contamination (default: 25)
CLUSTERING PARAMETERS:
-pa P_ANI, --P_ani P_ANI
ANI threshold to form primary (MASH) clusters
(default: 0.9)
-sa S_ANI, --S_ani S_ANI
ANI threshold to form secondary clusters (default:
0.99)
TAXONOMY:
–run_tax generate taxonomy information (Tdb) (default: False)
–tax_method {percent,max}
Method of determining taxonomy
percent = The most descriptive taxonimic level with at least (per) hits
max = The centrifuge taxonomic level with the most overall hits (default: percent)
I/O PARAMETERS:
-g [GENOMES [GENOMES …]], --genomes [GENOMES [GENOMES …]]
genomes to cluster in .fasta format; can pass a number
of .fasta files or a single text file listing the
locations of all .fasta files (default: None)
–checkM_method {lineage_wf,taxonomy_wf}
Either lineage_wf (more accurate) or taxonomy_wf
(faster) (default: lineage_wf)
–genomeInfo GENOMEINFO
location of .csv file containing quality information
on the genomes. Must contain: [“genome”(basename of
.fasta file of that genome), “completeness”(0-100
value for completeness of the genome),
“contamination”(0-100 value of the contamination of
the genome)] (default: None)
参考
dRep doc
Are these microbes the same?
-
Konstantinidis, K. T., Ramette, A. & Tiedje, J. M. The bacterial species definition in the genomic era. Philos. Trans. R. Soc. B Biol. Sci. 361, 1929–1940 (2006).
-
Goris, J. et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91 (2007).
-
Richter, M. & Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. 106, 19126–19131 (2009).
-
Varghese, N. J. et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771 (2015).
-
Kim, M., Oh, H.-S., Park, S.-C. & Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351 (2014).
-
Luo, C. et al. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc. Natl. Acad. Sci. 108, 7200–7205 (2011).
-
Kashtan, N. et al. Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science 344, 416–420 (2014).
-
Snitkin, E. S. et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci. Transl. Med. 4, 148ra116–148ra116 (2012).
-
Chase, J. et al. Geography and Location Are the Primary Drivers of Office Microbiome Composition. mSystems 1, (2016).
-
Olm, M. R. et al. Identical bacterial populations colonize premature infant gut, skin, and oral microbiomes and exhibit different in situ growth rates. Genome Res. gr-213256 (2017).
-
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: A tool for fast and accurate genome de-replication that enables tracking of microbial genotypes and improved genome recovery from metagenomes. bioRxiv (2017). doi:10.1101/108142
IF: NA NA NA